Thalassemia is a disease of the blood in which there is increased destruction (hemolysis) of the red cells. The condition causes the body to produce abnormal haemoglobin red blood cells, which in turn causes anaemia. The patients of thalassemia have severe anaemia, which needs regular blood transfusions for treatment. Red blood cells are important for carrying oxygen around the body.
The type of thalassemia a person may have is down to how many faulty genes they have inherited.
In Alpha thalassemia, having one faulty gene will cause little or no effect to a person. Two faulty genes are associated with mild anaemia. Three mutated genes result in haemoglobin H disease that needs regular blood transfusions to treat chronic anaemia. Unborn babies with four faulty genes are unlikely to survive pregnancy.
Beta thalassemia also has different forms: beta thalassemia major, also called BTM, requires lifelong regular blood transfusions. Beta thalassemia intermediate is also known as BTI or non-transfusion dependent thalassemia or NTDT. This is a milder form of the condition and the severity will differ between individuals, from mild anaemia to the need for regular blood transfusions.
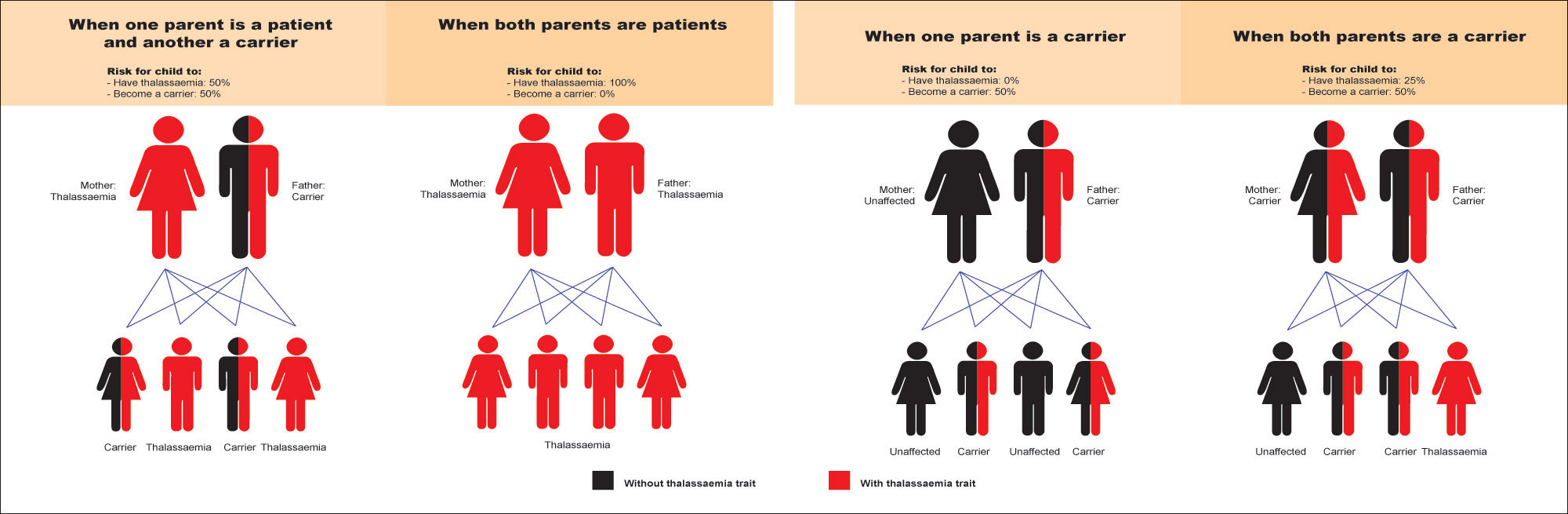
The thalassemia are inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Heterozygotes (i.e., carriers) may be slightly anaemic but are clinically asymptomatic. Carriers are often referred to as having thalassemia minor (or β-thalassemia minor). Carrier testing for individuals at risk (including family members, gamete donors, and members of at-risk ethnic groups) is possible. Once both HBB pathogenic variants have been identified in a couple at risk, prenatal testing and pre-implantation genetic diagnosis are possible.